HIV整合酶抑制剂Dolutegravir: 药物化学路线优化

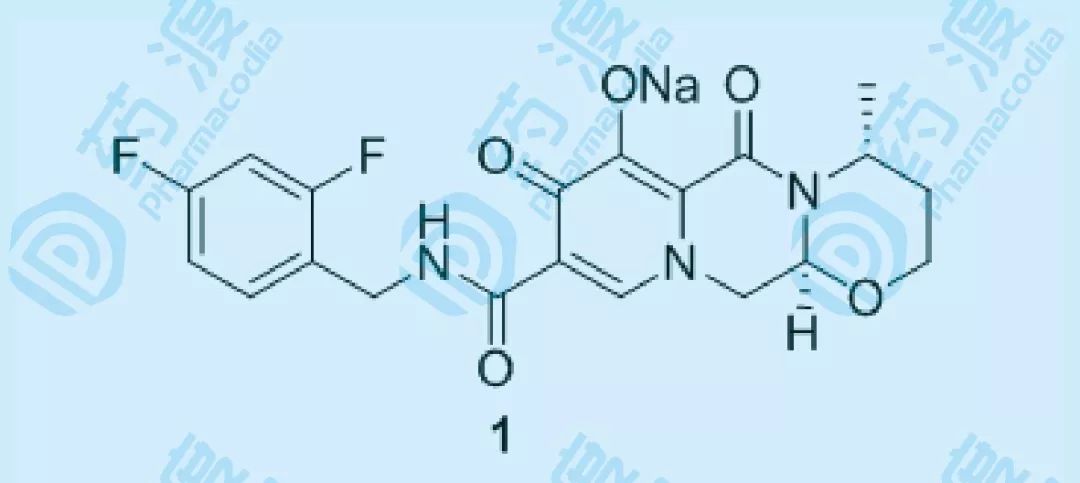
图一 Dolutegravir
Dolutegravir sodium(度鲁特韦)是新一代HIV-1治疗药物,通过抑制HIV病毒的整合酶达到抗病毒的作用,2013年8月13日获得FDA上市许可。Dolutegravir疗效显著,相比其他类型的抗HIV药物,由于人体内不含有整合酶,因此副作用少,对于HIV患者来说,是莫大的福音。
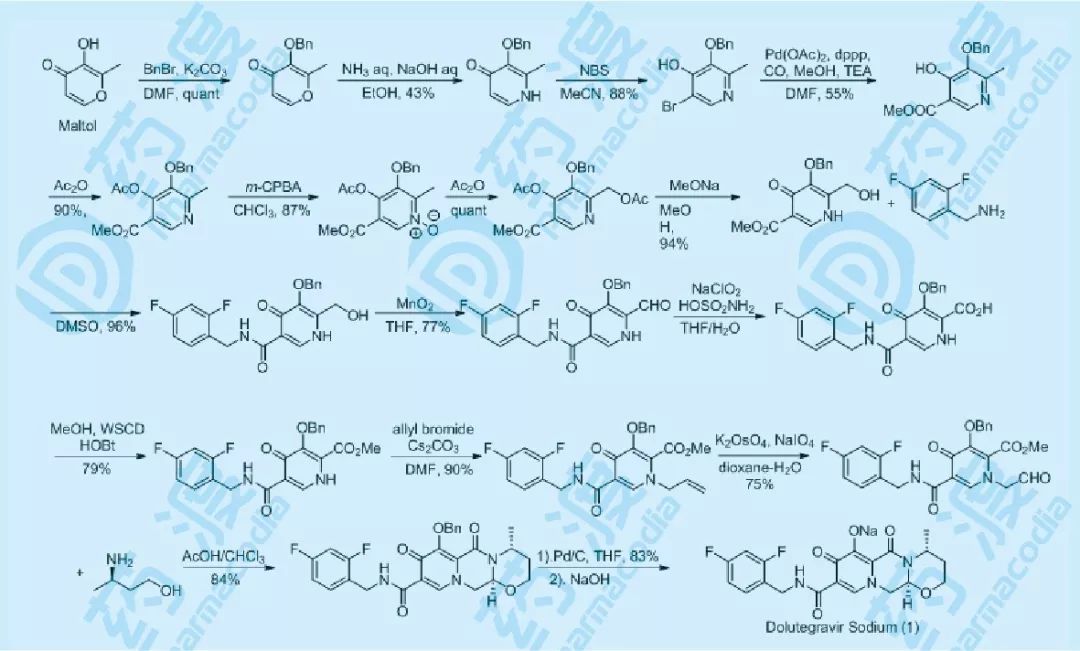
图二 Dolutegravir药物化学合成路线
在早期的开发中,Shionogi的药物化学家设计了如图所示的合成路线,一共17步反应,总收率约2%左右,其中的纯化均需要用到硅胶柱层析,这样的制备路线,仅能满足药物化学开发阶段的需要,在毒理以及早期的动物试验项目中,需求量更大,必须要有一条便于生产的路线,才能满足新药的继续开发。
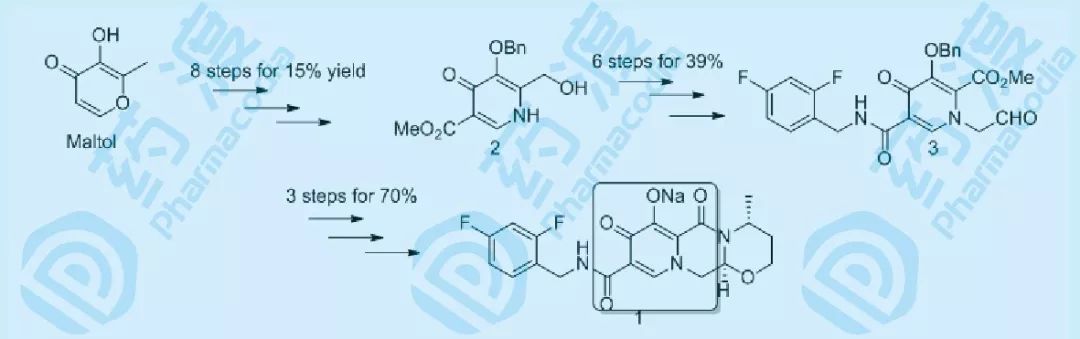
图三 合成路线分析
将上述药物化学路线分解成若干关键中间体,其中中间体2以麦芽酚(Maltol)作为起始原料,需要八步反应,总收率仅有15%。以多取代吡啶酮2出发,再经6步修饰以39%的收率得到前体化合物3,然后经过环化、脱保护等三步操作,以70%的收率得到最终产物,路线之冗余,效率之低!分析原因,大部分的操作都用在了构建多取代吡啶酮结构上,因此在进行优化设计时,首先要解决的问题就是关键中间体多取代吡啶酮的制备。
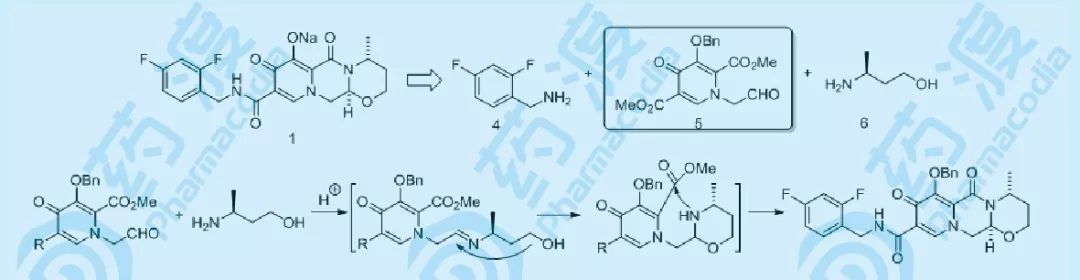
图四 工艺路线设计分析
根据对药物化学路线的分析,将1拆分为三个主要合成砌块:氟代苯乙胺4、多取代吡啶酮5以及手性氨基醇6,其中砌块5为关键的合成模块,5与4可通过很常见的酰化反应组合,而与6的组合,可采用药物化学路线中已经比较成熟的酸催化串联反应(机理如图四所示)。
多取代吡啶酮5的制备
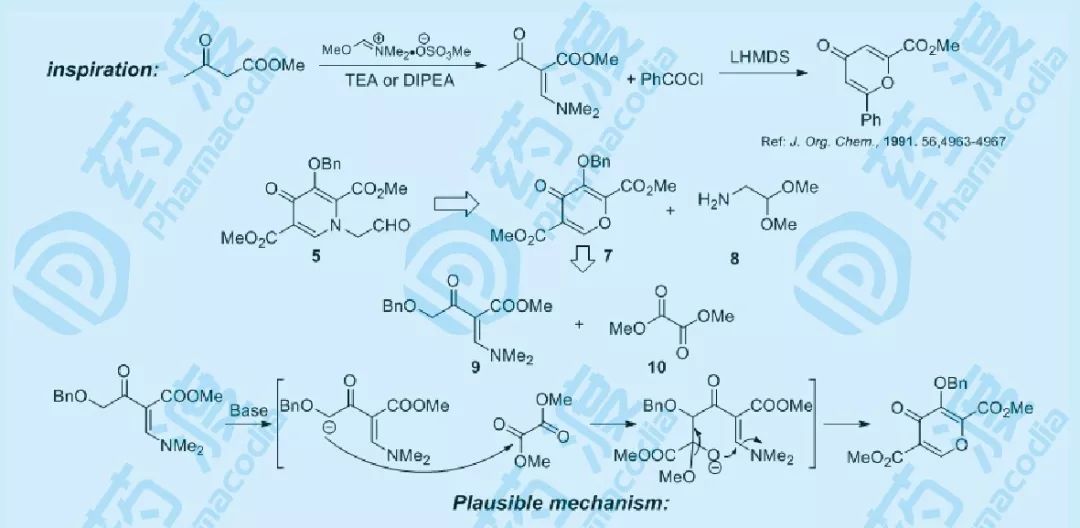
图五 关键中间体5的合成设计与机理分析
1991年,先灵葆雅公司的化学家Stuart W. McCombie报道了采用乙酰乙酸甲酯制备多取代吡喃的方法,而多取代吡啶化合物5则可以由多取代吡喃7与伯胺8在酸催化下得到。参照McCombie的方法,以廉价易得的9和10为起始原料,经inter-intramolecule的亲和加成-消除反应,可快速构建所需官能化的吡喃化合物7。
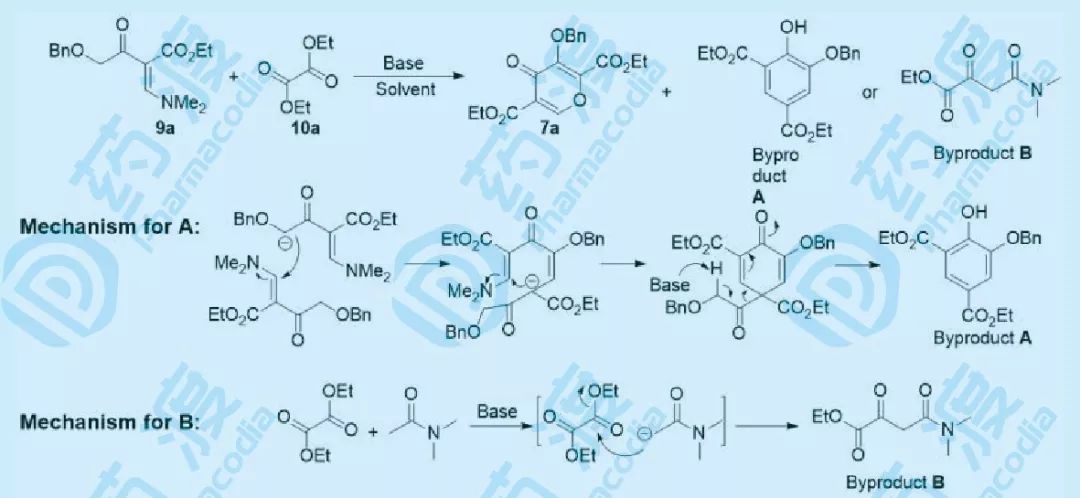
图六 多取代吡喃酮反应分析
根据上述设计路线,对多取代吡喃酮的构建进行了仔细的反应分析。最初在THF中,以NaH做碱,先加入9a,然后再加入10a,发现生成较多的副产物A,主要是原料9a发生了自身之间的反应(机理如图六),换用其他碱,如NaOtBu、KOtBu等,反应情况未得到改善,最好的收率仅能得到19%(NaH)。将溶剂由THF调整为DMA,发现有较多的副产物B生成(DMA与草酸二乙酯发生亲核加成-消去反应),由此分析可知,该反应需要在不含酸性质子的非质子极性溶剂中进行,如DMI。在对碱和溶剂进行优化未能得到满意的效果后,调整加料顺序,将9a和10a的混合物加入到碱溶液中,发现收率较先前的加料方式有所提高,表明9a能够快速在碱的作用发生去质子化,然后迅速与10a发生反应,改用新的加料顺序并调整10a的当量后,收率得到极大的提高。
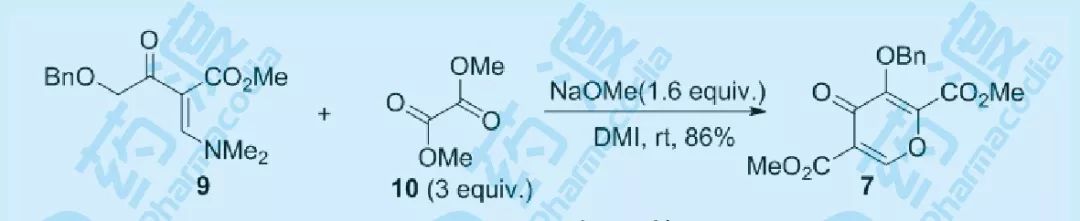
图七 多取代吡喃酮反应优化结果
经过前期的探索,将9和10的混合物加入到NaOtBu的DMI溶液中,将10的用量增加到3当量,用1.5当量的碱,能够以86%的收率得到目标产物7,并且未观察到酯交换产物的出现,考虑到NaOtBu的价格因素,将其替换为更为便宜的NaOMe,仍然能够以高收率得到目标产物。
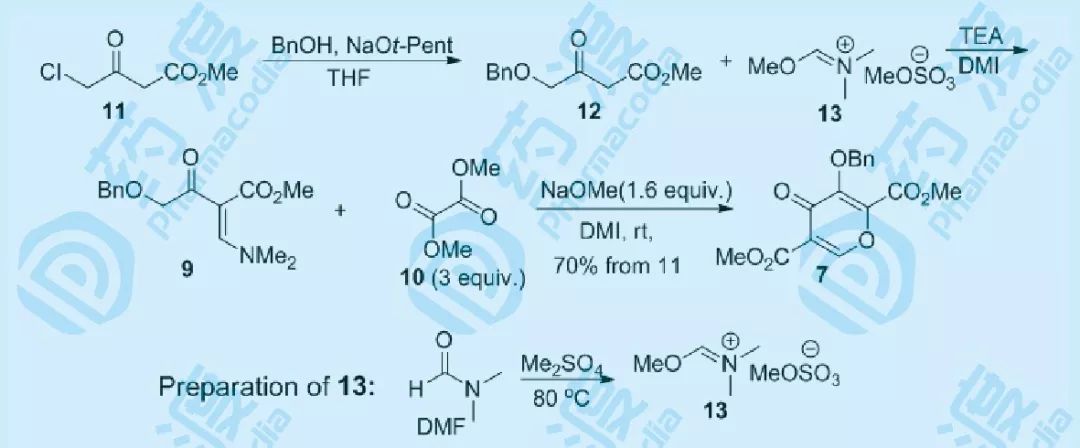
图八 多取代吡喃酮的制备路线
经过不断优化和尝试,7最终的制备路线如上:氯代物11在THF中与BnOH在叔戊醇钠(NaOt-Pent)作用下,得到12(NaOtBu会产生相当部分的叔丁醚产物),而13则由DMF与Me2SO4制备。将12与13在DMI中反应得到9,然后在优化后的条件下可顺利得到7,并由结晶析出纯化,三步收率为70%,相比药物化学路线8步反应15%的收率有了相当大的提高。
Dolutegravir sodium的合成
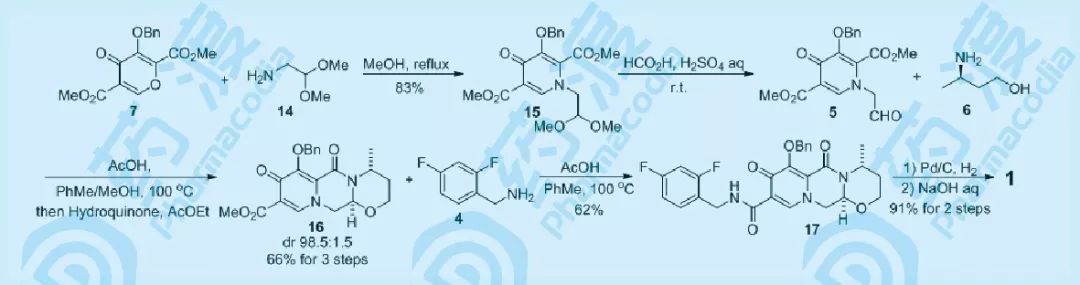
图九 砌块的组合
顺利解决多取代吡喃化合物7的制备问题后,参照先前药物化学路线中的方法,将7与14在甲醇中回流,以83%的结晶收率得到吡啶酮15,15在酸性水溶液的作用下脱除缩醛保护,然后与手性氨基醇6在醋酸作用下环合,然后溶于乙酸乙酯中并加入对苯二酚,产物16与1/2对苯二酚形成共结晶物析出(dr: 98.5:1.5, 三步收率66%),将含有1/2对苯二酚的16与伯胺4在AcOH催化下发生胺酯交换反应,以62%的收率得到17,再经脱保护、钠盐化(两步91%)得到最终目标产物Dolutegravir sodium。
经过优化后,新路线一共经9步反应,以22%的总收率得到最终产物Dolutegravir,相比药物化学路线17步、2%的总收率有了很大的提高,而且整个反应路径中不涉及氧化态的调整,原子经济性高,纯化过程简单。但其中关键中间体16的非对映选择性还不能达到API的质量要求(dr: 98.5:1.5),后续的几步操作收率仍然不够理想,因此还需要进一步的优化调整。
参考资料:
1.Practical Synthetic Method for the Preparation of Pyrone Diesters: An Efficient Synthetic Route for the Synthesis of Dolutegravir Sodium.(DOI: 10.1021/acs.oprd.8b00410).
2.Generation and in Situ Acylation of Enaminone Anions: A Convenient Synthesis of 3-Carbethoxy-4(1H)-pyridinones and -4-pyrones and Related Compounds. J.Org. Chem. 1991,56,4963-4967.
3.Carbamoyl Pyridone HIV‑1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles.J. Med. Chem. 2013, 56,1124−1135.

终轮通知||Gerhard Levy药动/药效(PK/PD)课程



立即解锁你的掌上专业工具!

扫码实时看更多精彩文章



